


Superfund Research Program
Cadmium-Linked Inflammation Increases the Severity of Lung Infection
View Research Brief as PDF(9.2MB)
Release Date: 07/12/2023
![]() subscribe/listen via iTunes, download(6.9MB), Transcript(112KB)
subscribe/listen via iTunes, download(6.9MB), Transcript(112KB)
Researchers funded in part by the NIEHS Superfund Research Program (SRP) uncovered a key mechanism explaining how inflammation caused by cadmium exposure makes lung infections more severe and deadly.
Fine particulate matter in air pollution often includes cadmium, a harmful heavy metal emitted from smelters and coal fired plants, for example. Cadmium exposure has previously been linked with higher risk of lung disease and death from flu and bacterial pneumonia infections, but the underlying mechanism involved was not known.
To explore immune changes that may explain this phenomenon, scientists led by Brent Carter, M.D., at the University of Alabama, Birmingham SRP Center, studied mice exposed to cadmium and Streptococcus pneumoniae — bacteria that account for more than half of the 1.4 million global annual deaths caused by lower respiratory tract infections.
Specifically, they were interested in the function of macrophages, important immune cells that develop in the bone marrow as a type of white blood cell called a monocyte. Monocytes are recruited to the lungs during infection where they become macrophages that first stimulate inflammation to defend against pathogens, and then down-regulate inflammation as the pathogen is neutralized. This process helps to repair the injury that occurred during the infection.
Pinpointing Problems with PPAR-Gamma
Mice were treated with either cadmium chloride or a saline placebo alone, or followed by Streptococcus pneumoniae infection five days later. Saline or cadmium exposure alone had no effect on survival, however, adding S. pneumoniae infection decreased survival by 20% and more than 50%, respectively.
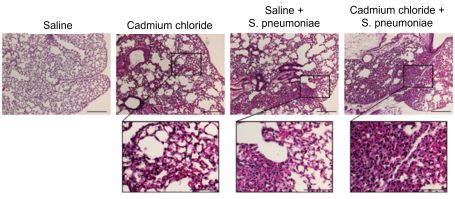
Cadmium-exposed mice had more macrophages in their lungs and evidence of lung injury compared to saline-exposed mice, a difference that was even more pronounced after S. pneumoniae infection.
Using single-cell RNA sequencing, they found that PPAR-gamma was predominantly expressed in macrophages compared to other cells in the lungs. PPAR-gamma is a transcription factor that normally helps macrophages reduce inflammation by blocking the production of other molecules involved in inflammation and oxidative stress. PPAR-gamma expression was similar in macrophages exposed to cadmium alone, saline and S. pneumoniae, or to cadmium and S. pneumoniae in combination.
Exposure to both cadmium and S. pneumoniae increased the production of pro-inflammatory TNF-alpha and interleukin-6, which PPAR-gamma normally inhibits, and decreased production of the anti-inflammatory interleukin-10, which PPAR-gamma normally stimulates.
These changes were explained by the team’s finding that exposure to cadmium and S. pneumoniae activated extracellular signal-regulated kinase (ERK) — a signaling molecule that plays important roles in immune response — in recruited macrophages. ERK triggered changes to PPAR-gamma that caused it to degrade, thus canceling its anti-inflammatory role.
Translating to Humans and Targeting Treatments
To evaluate the human relevance of their findings, the researchers examined residents living in an industrial area of North Birmingham, Alabama, and found they had increased cadmium levels in their lung fluid compared to those from a control area with no industrial activity.
Higher cadmium levels were associated with a loss of epithelial barrier function among study participants, a condition that can make it easier for blood proteins to leak into the lung fluid, a sign of lung injury. Compared to the control group, North Birmingham residents also showed inhibition of PPAR-gamma, ERK activation, and similar trends of increased TNF-alpha and interleukin-6 and reduced levels of interleukin-10.
Finally, the team conducted studies in mice to test the ability of a drug called BVD-523, an ERK inhibitor that is currently in clinical trials as a cancer treatment, to protect against lung damage. BVD-523 blocked ERK activation and subsequent PPAR-gamma degradation. Mice treated with BVD-523 did not exhibit the characteristic increase in TNF-alpha and interleukin-6 or decreased interleukin-10 following cadmium exposure or S. pneumoniae infection. More importantly, they were protected from lung injury and had better survival than mice not treated with BVD-523.
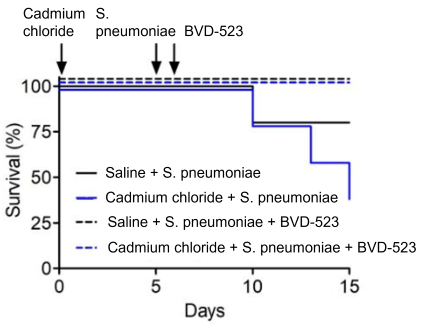
According to the authors, targeting regulation of PPAR-gamma in monocyte-derived macrophages via ERK could be a novel target to reduce the severity of lung injury following exposure to air pollution and respiratory infections.
For More Information Contact:
A. Brent Carter
University of Alabama at Birmingham
Division of Pulmonary, Allergy and Critical Care Medicine
421 THT
Birmingham, Alabama 35294-0006
Phone: 205-934-1682
Email: bcarter1@uab.edu
To learn more about this research, please refer to the following sources:
- Larson-Casey JL, Liu S, Pyles JM, Lapi SE, Saleem K, Antony V, Gonzalez ML, Crossman DK, Carter AB. 2023. Impaired PPARy activation by cadmium exacerbates infection-induced lung injury. JCI Insight 8(9):e166608. doi:10.1172/jci.insight.166608 PMID:36928191 PMCID:PMC10243824
To receive monthly mailings of the Research Briefs, send your email address to srpinfo@niehs.nih.gov.